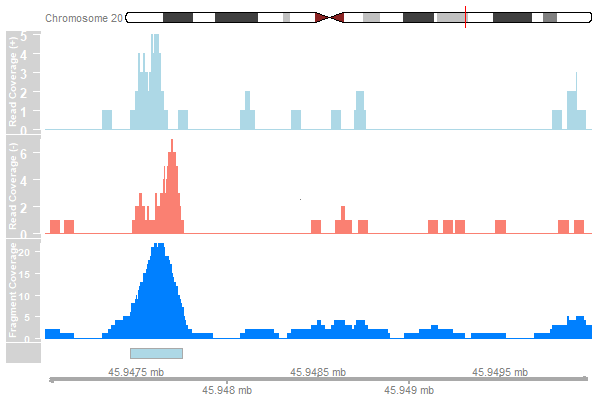
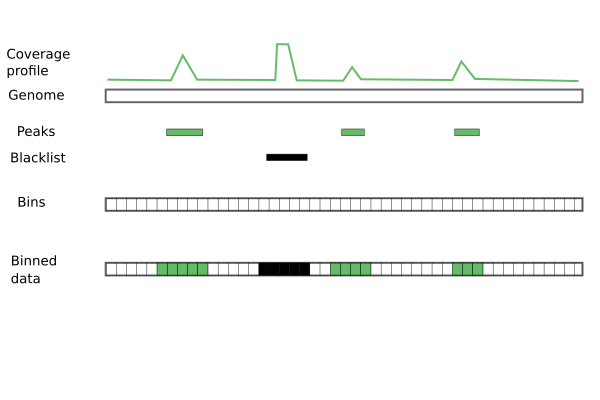
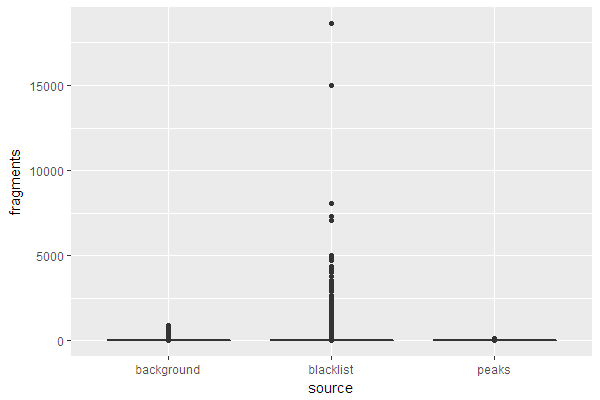
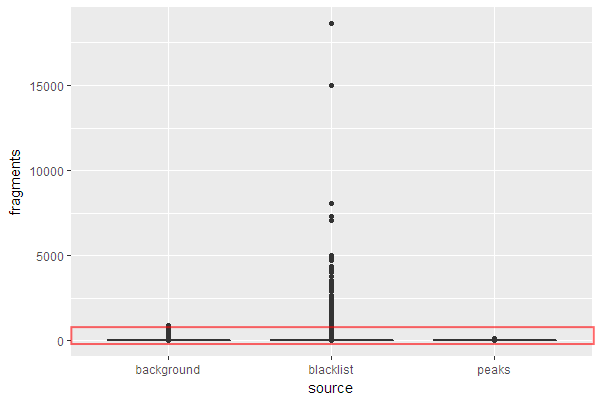
count_bins <- function(reads, target, bins){
# Find all bins overlapping peaks
overlap <- from(findOverlaps(bins, target))
target_bins <- bins[overlap, ]
# Count the number of reads overlapping each peak bin
target_bins$score <- countOverlaps(target_bins, reads)
target_bins
}