Interpreting ChIP-seq peaks
ChIP-seq with Bioconductor in R

Peter Humburg
Statistician, Macquarie University
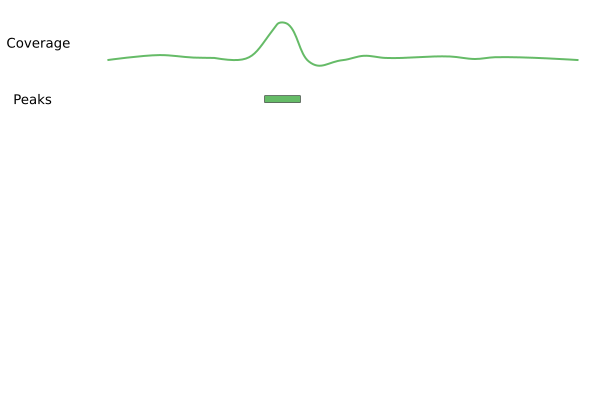
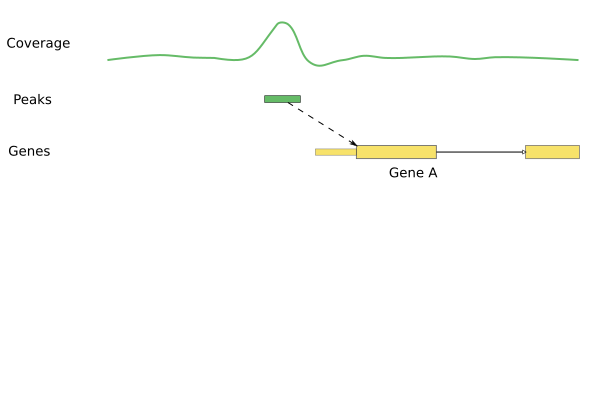
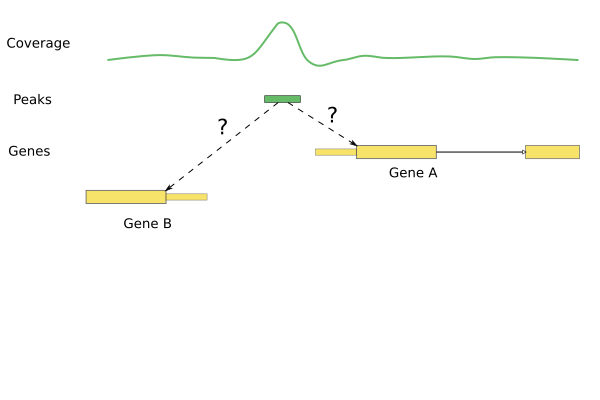
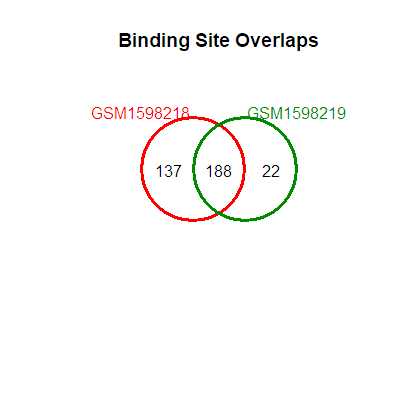
Visualizing similarities and differences
library(DiffBind)
dba.plotVenn(peaks, mask=1:2)
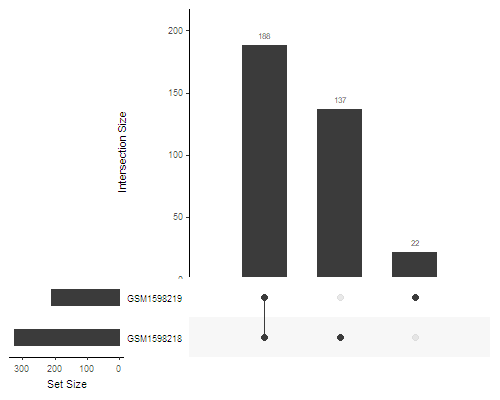
Using UpSet plots
library(UpSetR)
called_peaks <- as.data.frame(peaks$called)
upset(called_peaks, sets=colnames(peaks$called), order.by='freq')