ChIP-seq with Bioconductor in R
Peter Humburg
Statistician, Macquarie University
SRR1782620.7265769
0
chr20 29803915
51M
0 0
AATGAAATGGAA
CCCFFFFFHHHH
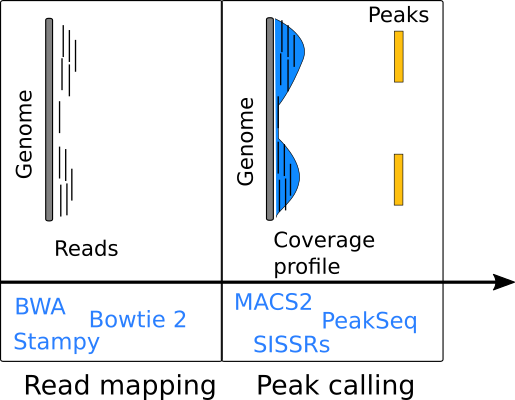
Rsamtools
Use readGAlignments to import mapped reads.
readGAlignments
library(GenomicAlignments) reads <- readGAlignments(bam_file)
Returns GAlignments object.
GAlignments
BamViews
library(GenomicRanges) library(Rsamtools) ranges <- GRanges(...) views <- BamViews(bam_file, bamRanges=ranges)
reads <- readGAlignments(views)
The BamViews function supports multiple BAM files.
Use import.bed to load peak calls from a BED file.
import.bed
library(rtracklayer) peaks <- import.bed(peak_bed, genome="hg19")
Use peaks to define views into the BAM files.
peaks
bams <- BamViews(bam_file, bamRanges=peaks) reads <- readGAlignments(bams)