ChIP-seq Review
ChIP-seq with Bioconductor in R

Peter Humburg
Statistician, Macquarie University
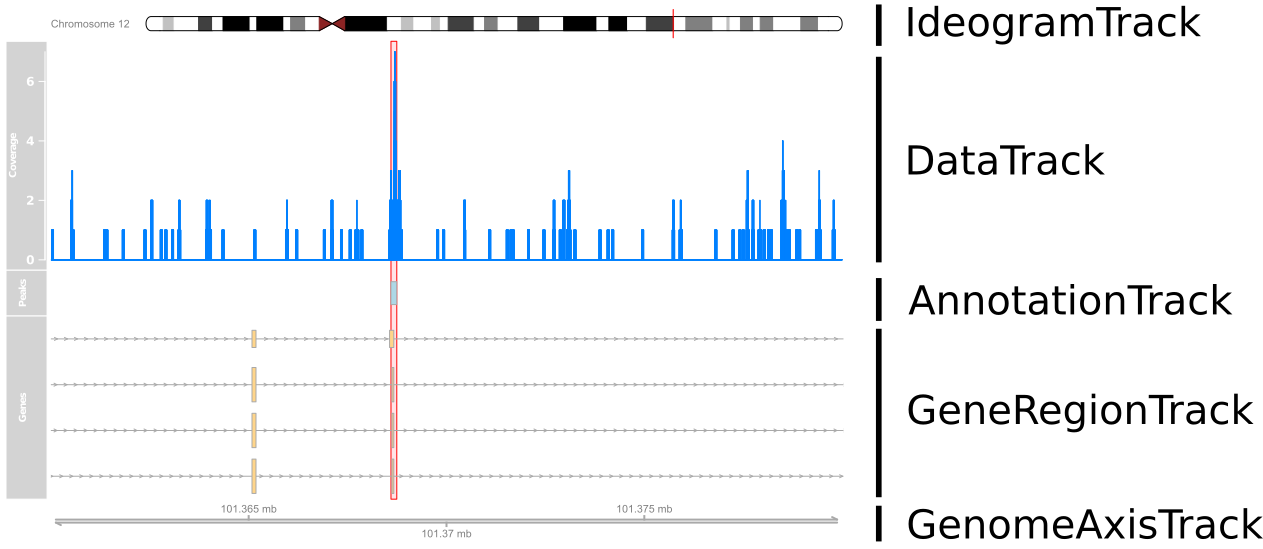
Review: Visualising ChIP-seq data
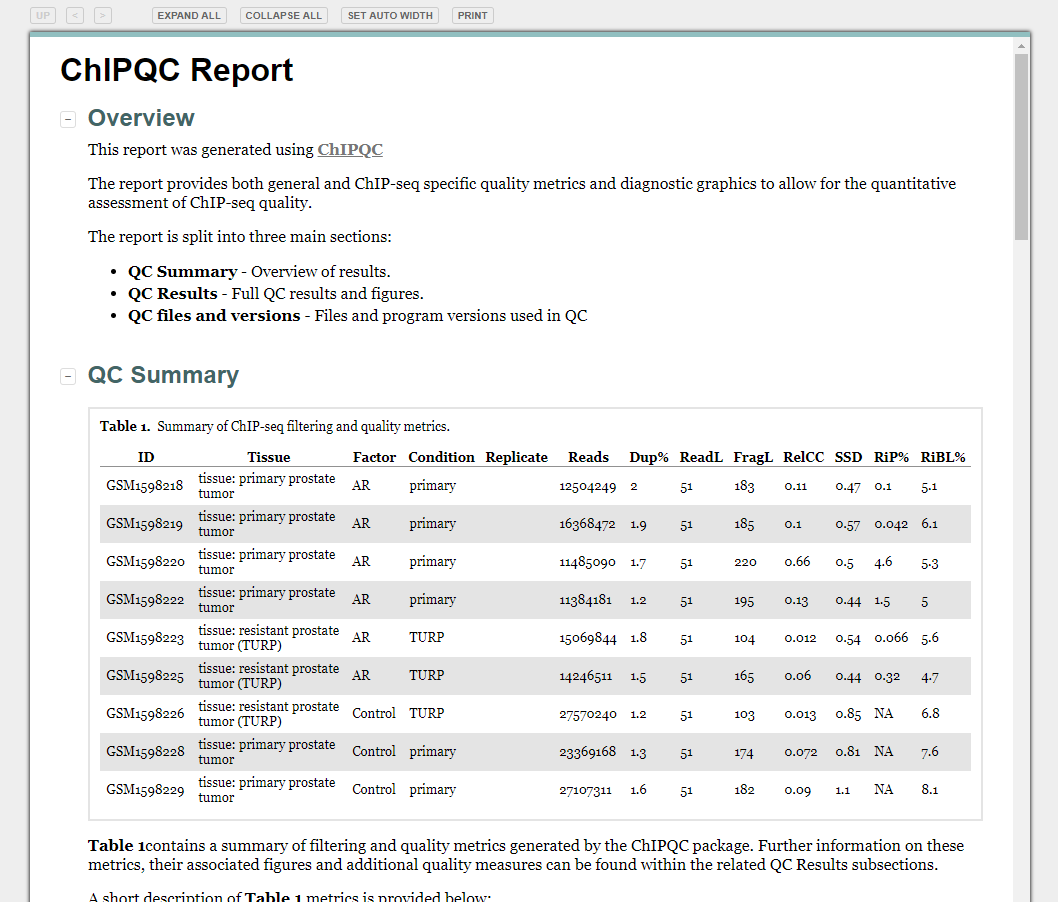
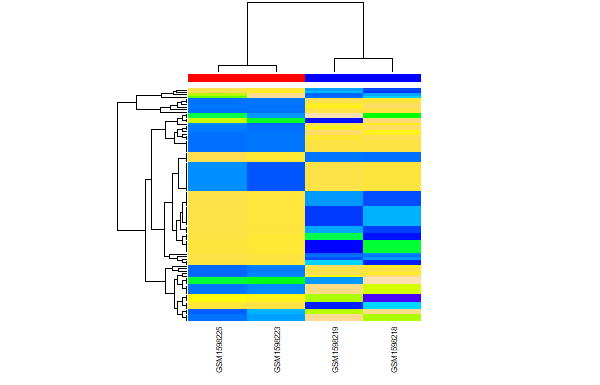
Review: Differential Binding
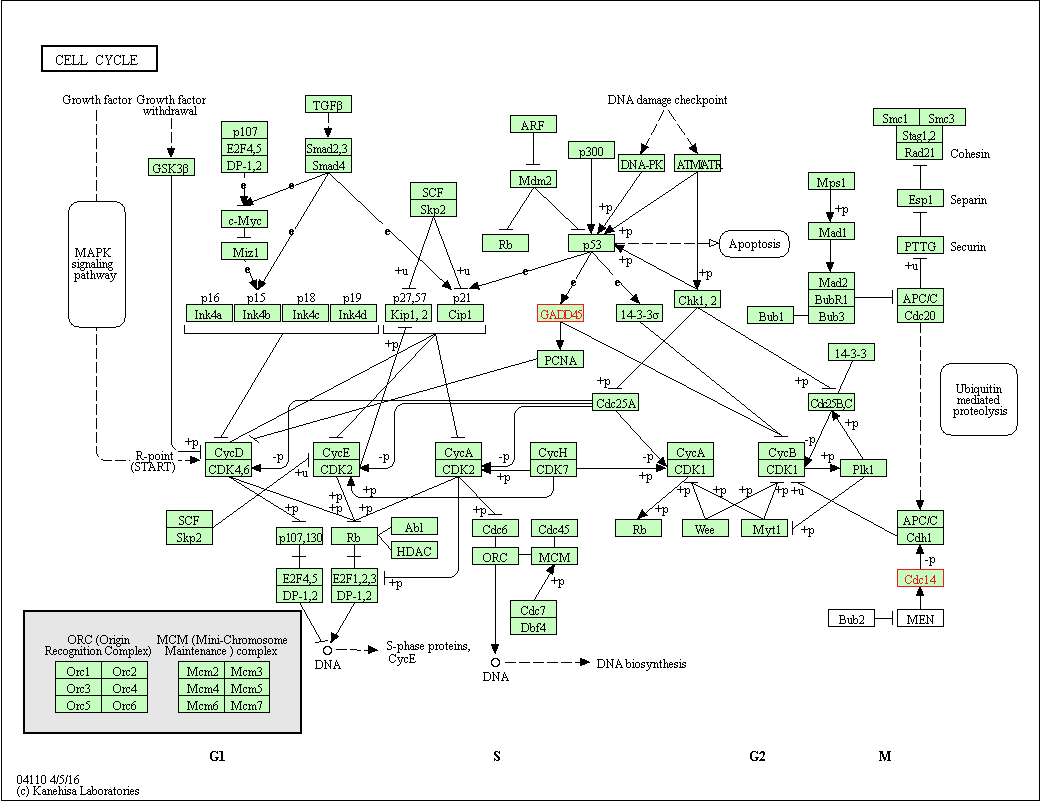
Review: Enrichment analysis
ChIP-seq with Bioconductor in R
Peter Humburg
Statistician, Macquarie University

