Knitr, dplyr, markdown, ggplot2, digest, shiny, and Rcpp
rqcQA
library(Rqc)
files <- # get the full path of the files you want to assess
qaRqc <- rqcQA(files)
# exploring qaRqc
class(qaRqc) # "list"
names(qaRqc) # name of the input files
# for each file
qaRqc[1]
# the class of the results is RqcResultSet
rqcQA arguments
library(Rqc)
# get the path of the files you want to assess
files <- "data/seq1.fq" "data/seq2.fq" "data/seq3.fq" "data/se4.fq"
qaRqc <- rqcQA(files, workers = 4)
# sample of sequences
set.seed(1111)
qaRqc_sample <- rqcQA(files, workers = 4, sample = TRUE, n = 500))
# paired-end files
pfiles <- "data/seq_11.fq" "data/seq1_2.fq" "data/seq2_1.fq" "data/seq2_2.fq"
qaRqc_paired <- rqcQA(pfiles, workers = 4, pair = c(1, 1, 2, 2)))
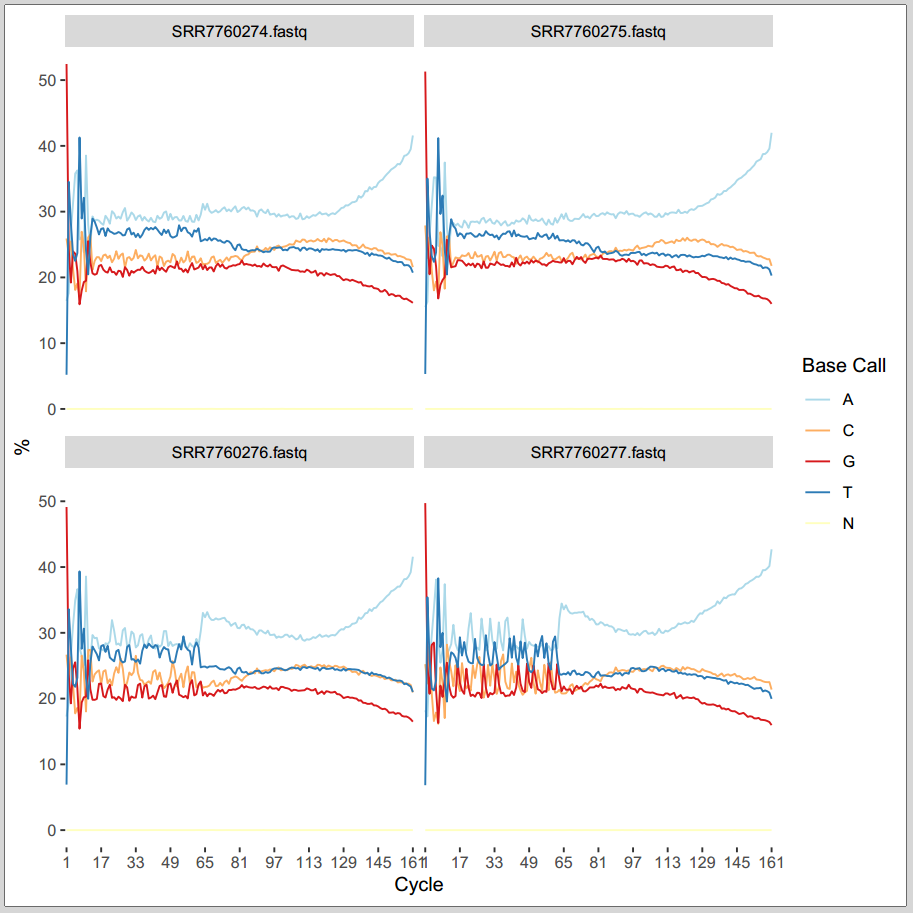
rqcReport and rqcResultSet
# create a report
reportFile <- rqcReport(qaRqc, templateFile = "myReport.Rmd")
browseURL(reportFile)
#The class of qaRqc is rqcResultSet
methods(class = "RqcResultSet")