Gene of interest
Introduction to Bioconductor in R

Paula Andrea Martinez, PhD.
Data Scientist
Examples of genomic intervals
- Reads aligned to a reference
- Genes of interest
- Exonic regions
- Single nucleotide polymorphisms (SNPs)
- Regions of transcription or binding sites, RNA-seq or ChIP-seq
Genomic Ranges
library(GenomicRanges)
(myGR <- GRanges("chr1:200-300"))
GRanges object with 1 range and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
[1] chr1 [200, 300] *
-----
seqinfo: 1 sequence from an unspecified genome; no seqlengths
- GRanges class is a container to save genomic intervals by chromosome
- Minimum arguments
chr1:200-300 - GRanges
seqnamesandseqinfo
# df a data.frame like structure
seqnames start end strand score GC
1 chrX 50 120 + 1 0.25
2 chrX 130 140 + 2 0.25
3 chrX 153 154 + 3 0.25
4 chrY 30 40 * 4 0.25
5 chrY 50 55 - 5 0.25
(myGR <- as(df, "GRanges")) # transform df into GRanges
GRanges object with 5 ranges and 2 metadata columns:
seqnames ranges strand | score GC
<Rle> <IRanges> <Rle> | <integer> <numeric>
[1] chrX [ 50, 120] + | 1 0.25
[2] chrX [130, 140] + | 2 0.25
[3] chrX [153, 154] + | 3 0.25
[4] chrY [ 30, 40] * | 4 0.25
[5] chrY [ 50, 55] - | 5 0.25
-----
seqinfo: 2 sequences from an unspecified genome; no seqlengths
Genomic Ranges accessors
methods(class = "GRanges") # to check available accessors# used for chromosome names seqnames(gr)# returns an IRanges object for ranges ranges(gr)# stores metadata columns mcols(gr)# generic function to store sequence information seqinfo(gr)# stores the genome name genome(gr)
- Accessors are both setter and getter functions
- Accessors can be inherited thanks to S4 definitions
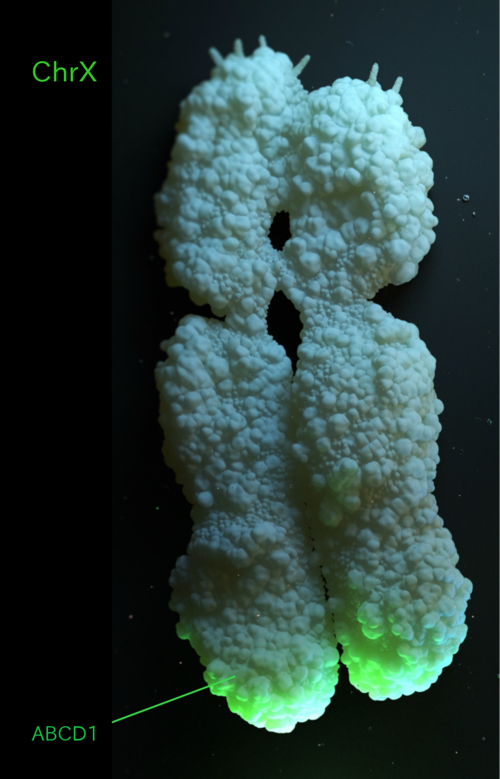
Gene of interest: ABCD1
- ABCD1 is located at the end of chromosome X long arm
- encodes a protein relevant for the well functioning of brain and lung cells in mammals
- chrX is ~ 156 mi bp
- Located chrX ~ 153.70 mi bp
Chromosome X GRanges
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
hg <- TxDb.Hsapiens.UCSC.hg38.knownGene
Select genes from chromosome X
hg_chrXg <- genes(hg, filter = list(tx_chrom = c("chrX")))
GRanges object with 1192 ranges and 1 metadata column:
seqnames ranges strand | gene_id
<Rle> <IRanges> <Rle> | <character>
100008586 chrX 49551278-49568218 + | 100008586
10009 chrX 120250752-120258398 + | 10009
100093698 chrX 13310652-13319933 + | 100093698
... ... ... ... . ...
-------
seqinfo: 640 sequences (1 circular) from hg38 genome
Let's practice looking for a gene of interest in the human genome!
Introduction to Bioconductor in R

