Manipulating collections of GRanges
Introduction to Bioconductor in R

Paula Andrea Martinez, PhD.
Data Scientist
GRangesList
- The
GRangesList-classis a container for storing a collection ofGRanges- Efficient for storing a large number of elements.
- To construct a
GRangesListas(mylist, "GRangesList")GRangesList(myGranges1, myGRanges2, ...)
- To convert back to
GRangesunlist(myGRangesList)
- Accessors
methods(class = "GRangesList")
When to use lists?
- Multiple GRanges objects may be combined into a GRangesList
- GRanges in a list will be taken as compound features of a larger object
- Examples of GRangesLists are
- transcripts by gene
- exons by transcripts
- read alignments
- sliding windows
# GRanges object with 983 genes hg_chrXslidingWindows(hg_chrX, width = 20000, step = 10000)
# showing only two elements of the list
GRangesList object of length 983:
[[1]]
GRanges object with 2 ranges and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
[1] chrX [276322, 296321] +
[2] chrX [286322, 303356] +
[[2]]
GRanges object with 3 ranges and 0 metadata columns:
seqnames ranges strand
[1] chrX [624344, 644343] +
[2] chrX [634344, 654343] +
[3] chrX [644344, 659411] +
...
GenomicFeatures uses transcript database (TxDb) objects to store metadata, manage genomic locations and relationships between features and its identifiers.
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
(hg <- TxDb.Hsapiens.UCSC.hg38.knownGene)
Db type: TxDb
Supporting package: GenomicFeatures
Data source: UCSC
Genome: hg38
Organism: Homo sapiens
Taxonomy ID: 9606
Resource URL: http://genome.ucsc.edu/
Type of Gene ID: Entrez Gene ID
transcript_nrow: 197782
exon_nrow: 581036
cds_nrow: 293052
Db created by: GenomicFeatures package from Bioconductor
Creation time: 2016-09-29 13:02:09 +0000 (Thu, 29 Sep 2016)
Genes, transcripts, exons
library(TxDb.Hsapiens.UCSC.hg38.knownGene) hg <- TxDb.Hsapiens.UCSC.hg38.knownGene # hg is a A TxDb objectseqlevels(hg) <- c("chrX") # prefilter results to chrX# transcripts transcripts(hg, columns = c("tx_id", "tx_name"), filter = NULL) # exons exons(hg, columns = c("tx_id", "exon_id"), filter = list(tx_id = "179161"))
columns and filter can be NULL or any of these:
"gene_id", "tx_id", "tx_name", "tx_chrom", "tx_strand",
"exon_id", "exon_name", "exon_chrom", "exon_strand",
"cds_id", "cds_name", "cds_chrom", "cds_strand" and "exon_rank"
Exons by transcripts
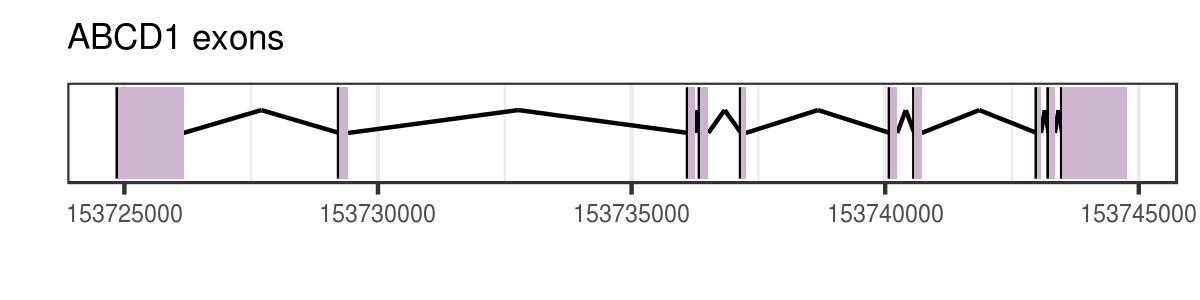
hg <- TxDb.Hsapiens.UCSC.hg38.knownGene seqlevels(hg) <- c("chrX") # prefilter chromosome X exonsBytx <- exonsBy(hg, by = "tx") # exons by transcriptabcd1_179161 <- exonsBytx[["179161"]] # transcript idwidth(abcd1_179161) # width of each exon, the purple regions of the figure
1299 181 143 169 95 146 146 85 126 1274
Overlaps
# countOverlaps results in an integer vector of counts
countOverlaps(query, subject)
# findOverlaps results in a Hits object
findOverlaps(query, subject)
# subsetByOverlaps returns a GRangesList object
subsetByOverlaps(query, subject)
- Query and subject are either a GRanges or GRangesList objects.
- Overlaps might be complete all partial.
It's your turn to put this into practice!
Introduction to Bioconductor in R

