Visualization of results
RNA-Seq with Bioconductor in R

Mary Piper
Bioinformatics Consultant and Trainer
Visualizing results - Expression heatmap
# Subset normalized counts to significant genes sig_norm_counts_wt <- normalized_counts_wt[wt_res_sig$ensgene, ]# Choose a color palette from RColorBrewer library(RColorBrewer) heat_colors <- brewer.pal(6, "YlOrRd")display.brewer.all()
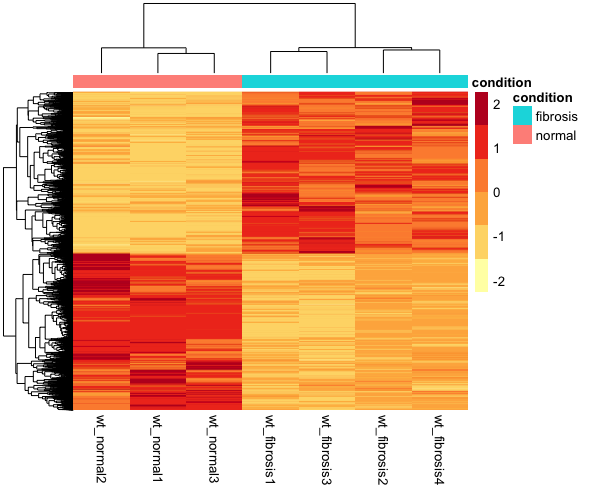
Visualizing results - Expression heatmap
# Run pheatmap
pheatmap(sig_norm_counts_wt,
color = heat_colors,
cluster_rows = T,
show_rownames = F,
annotation = select(wt_metadata, condition),
scale = "row")
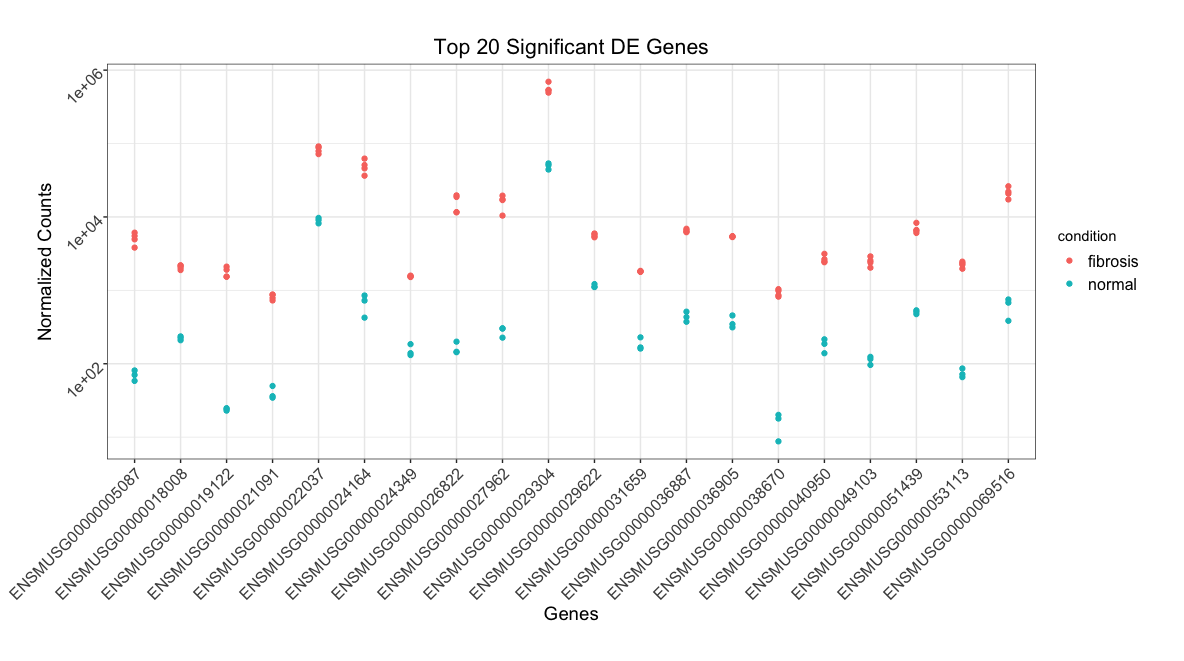
ggplot(wt_res_all) +
geom_point(aes(x = log2FoldChange, y = -log10(padj), color = threshold)) +
xlab("log2 fold change") +
ylab("-log10 adjusted p-value") +
ylim=c(0, 15) +
theme(legend.position = "none",
plot.title = element_text(size = rel(1.5), hjust = 0.5),
axis.title = element_text(size = rel(1.25)))
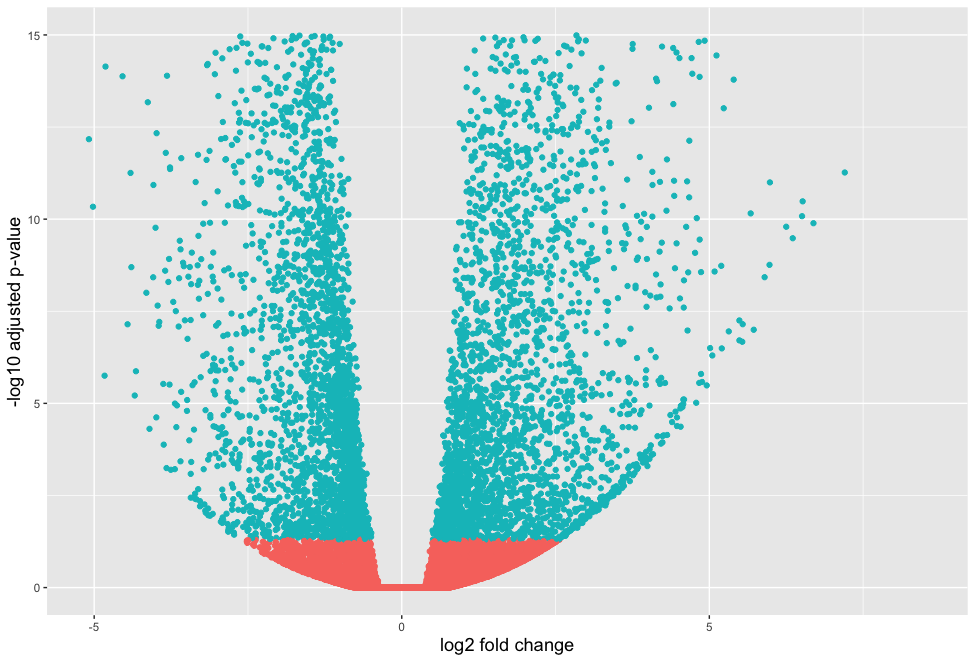
Visualizing results - Expression plot
Significant results:
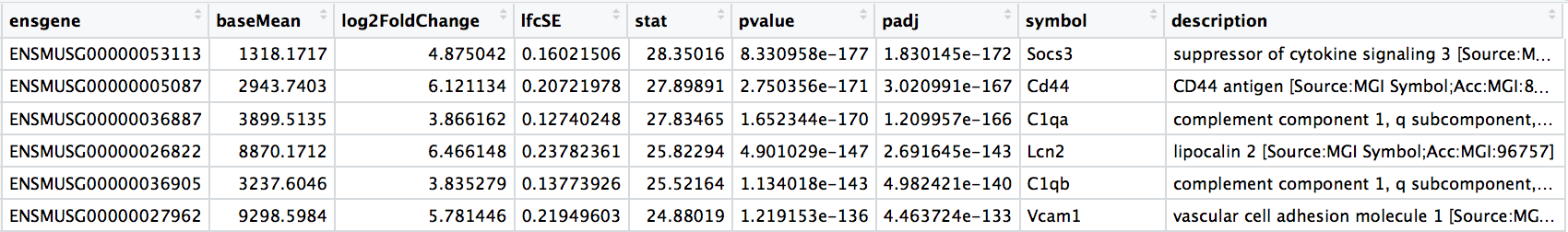
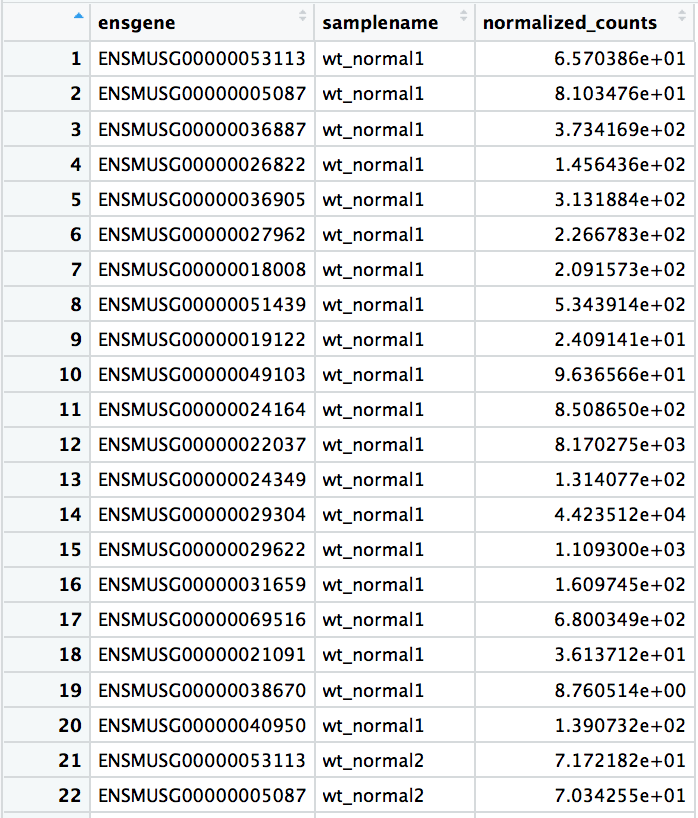
Normalized counts for significant genes:
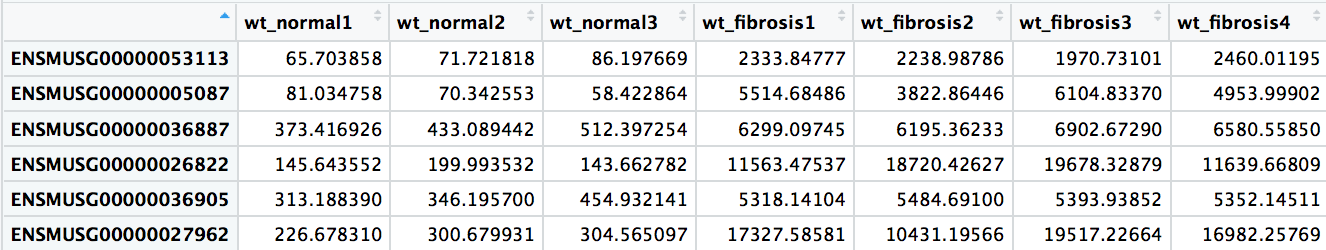
Visualizing results - Expression plot
top_20 <- data.frame(sig_norm_counts_wt)[1:20, ] %>% rownames_to_column(var = "ensgene")top_20 <- gather(top_20, key = "samplename", value = "normalized_counts", 2:8)