Organizing the data for DESeq2
RNA-Seq with Bioconductor in R

Mary Piper
Bioinformatics Consultant and Trainer
Bringing in data for DESeq2: sample order
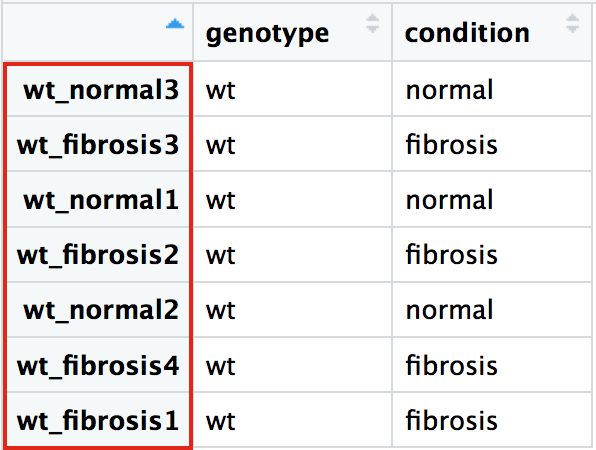
Metadata
Raw counts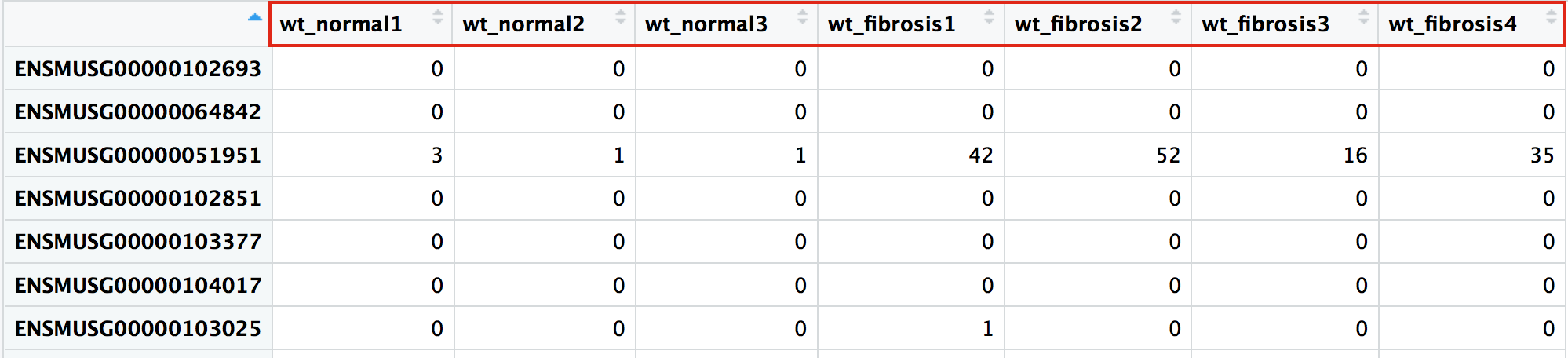
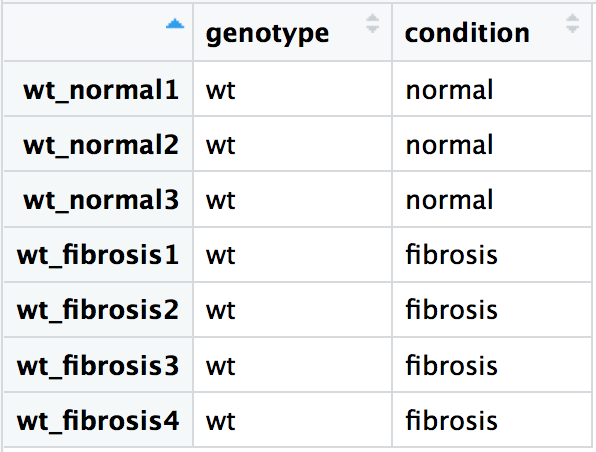
all(rownames(reordered_wt_metadata) == colnames(wt_rawcounts))
TRUE
Creating the DESeq2 object
# Create DESeq object
dds_wt <- DESeqDataSetFromMatrix(countData = wt_rawcounts,
colData = reordered_wt_metadata,
design = ~ condition)